
Journal of Korean Society of Dental Hygiene![]() Open access, Peer Reviewed
Open access, Peer Reviewed
pISSN 2287-1705, eISSN 2288-2294
Weeks in Review
Weeks to Publication
Journal of Korean Society of Dental Hygiene![]() Open access, Peer Reviewed
Open access, Peer Reviewed
pISSN 2287-1705, eISSN 2288-2294
Kyung-Hee Lee![]()
Department of Dental Hygiene, Dongseo University
Correspondence to Kyung-Hee Lee, Department of Dental Hygiene, Dongseo University, 47 Jurye-ro, Sasang-gu, Busan-si, 47011, Korea. Tel: +82-51-320-2730, Fax: +82-51-320-2752, E-mail: kyhee@dongseo.ac.kr
Volume 24, Number 3, Pages 219-27, June 2024.
J Korean Soc Dent Hyg 2024;24(3):219-27. https://doi.org/10.13065/jksdh.20240303
Received on May 13, 2024 , Revised on May 31, 2024, Accepted on June 05, 2024, Published on June 30, 2024.
Copyright © 2024 Journal of Korean Society of Dental Hygiene.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License(http://creativecommons.org/licenses/by-nc/4.0)

Objectives: The pulp is the center of the tooth containing nerves and blood vessels. The condition in which the pulp becomes inflamed due to caries or periodontitis is called pulpitis. Pulpitis is a difficult-to-treat disease and causes peripheral nerve tissue changes and severe pain; however, the relationship between neuronal activity and voltage-gated sodium channel 1.7 (Nav1.7) expression in the trigeminal ganglion (TG) during pulpitis has not been well studied. In this study, we found that experimentally induced pulpitis activates Nav1.7 expression in the periphery, leading to neuronal overexpression in the TG. Thus, we sought to identify ways to regulate this process. Methods: Acute pulpitis was induced in rat maxillary molars by treating the pulp with allyl isothiocyanate (AITC). Three days later, in vivo optical imaging was used to record and compare neural activities in the TG. Western blotting was used to identify molecular changes in terms of the expression of extracellular signal-regulated kinase (ERK), c-Fos, transient receptor potential ankyrin 1 (TRPA1), and collapsin response mediator protein-2 (CRMP2) in the brain stem. Results: The results confirmed the neurological changes in the TGs of the pulpitis model, and histological and molecular biological evidence confirmed that increased Nav1.7 expression induced by pulpitis leads to pain. Furthermore, selective inhibition of Nav1.7 resulted in changes in neural activity, suggesting that pulpitis induces increased Nav1.7 expression, and that effective control of Nav1.7 could potentially reduce pain. Conclusions: The inhibition of overexpressed Nav1.7 channels may modulate nociceptive signal processing in the brain and effectively control pain associated with pulpitis.
Neural activity, Nociceptive signal processing, Pulpitis pain, Sodium channel, Trigeminal ganglion
Pulpitis pain showed ectopic persistence and hyperalgesia as associated with pulp inflammation. Even though the dental pulpitis induced pain cause severe discomfort and affects patients’ quality of life, the mechanisms of pulpitis pain have not yet been fully elucidated and also the treatment strategies and long-term efficacy for dental pulpitis are relatively limited. The prostaglandin metabolites along with other inflammatory mediators in pulpitis may produce the accentuate pain by increasing peripheral and central sensitization [1]. The inflammatory mediators are important for both types of sensitizations and act on their corresponding receptors to activate intracellular signaling pathways.
The trigeminal nerve fiber is the primary nerve and predominantly innervates the orofacial region which is associated with the sodium channel, although there are various regions of orofacial pain origins. The trigeminal ganglia, sensation in the oral area, consists of sensory neuronal bodies of primary afferent nerve cells in the dental pulp [2] and is also involved in pain caused by most neuro-inflammation and hyperalgesia [3]. The stimuli are converted into electrical signals through molecular transducers called mechanical ion channels in dental primary afferent (DPA) neurons and odontoblasts [4]. In particular, transient receptor potential (TRP) ion channels have been found to be related to pulpitis pain transmission and are highly expressed in the trigeminal ganglia and on odontoblasts in the pulp [5]. It has been demonstrated that inflammatory mediators can decrease the activation threshold of transient receptor potential vanilloid type 1 (TRPV1), which can regulate the intracellular Ca2+ concentration [6]. Cha et al [7] analyzed the peripheral and central TRPV1 expressions following pulp inflammation. TRPV1 channel plays a crucial role in the peripheral transduction of noxious pulpitis pain, but not in the central nerve system (CNS). It is necessary to identify additional transducers that mediate signal transmission from pulpal afferents to their central targets.
Voltage-gated sodium channels (VGSCs), the electrical excitability of peripheral nerves, consist with fast-activating neurotoxin tetrodotoxin (TTX)-sensitive (Nav1.7) channels and slow-inactivating TTX-resistant channels [8]. Nav1.7 channels are deployed at nociceptor nerve terminals where generator potentials occur in response to stimulation of the sensory nerve endings. Nav1.7, preferentially expressed in dorsal root ganglion (DRG) neurons, plays a central role in inflammatory pain and nociceptive sensitization [9,10]. In the allyl isothiocyanate (AITC; Sigma-Aldrich, WI, USA) induced pulpitis model, Nav1.7 expression in the trigeminal ganglion (TG) was significantly higher than control and also suppressed the hyperpolarized activity by treatment of selective Nav1.7 blocker [11]. Nav1.7 due to its genetic links to pathological pain has need to fully elucidate the mechanisms underlying the contributions of these sodium channel isoforms in the induction and maintenance of pathological pain states. It was demonstrated a correlation between the expression levels of Nav1.7 and ERK and the degree of inflammatory pain by Nav1.7 inhibitor or an ERK inhibitor in TG treatment [12]. It is necessary to study the development of subtype-specific sodium channel blockers and understanding of the function of specific subtypes of sodium channels in the pathophysiology of inflammatory pain. And the molecular mechanisms of Nav1.7 trafficking to the neuronal membrane remained unclear in pulpitis pain.
The aim of this present study was to verify the exploitation of TG Nav1.7 blockers whether relief the hyperalgesia of inflammatory pulpitis pain by neural activity in vivo electrophysiology study. To investigate nociceptive signal processing changes by Nav1.7 channel blocking, we observed the nociceptor signaling differentiation in the central area of spinal trigeminal nucleus (SpVc) via western blot.
Male C57BL/6 mice (6 weeks old; 20–25 g; Orient Bio, Gyeonggi, Korea) were subjected to all experiments. These experiments were approved by the Institutional Animal Care and Use Committee (IACUC approval No. 2021-0173) in the Laboratory Animal Facility at Yonsei University (Seoul, Republic of Korea). The animals were housed in a 12-h light/dark cycle with food and water provided ad libitum. Mice were anesthetized using intraperitoneal (i.p.) sodium pentobarbital (75 mg/kg). Using a low-speed dental drill, the pulp of mouse’s the right maxillary first molar (M1) was gently exposed. For inflammation of tooth pulp, dental paper point (diameter, 0.15 mm; length, 20 mm) soaked in AITC was applied for 1 min in the exposed pulp. While saline was applied instead of AITC in the control group mice. The damage to the exposed M1 pulp cavity was sealed by visible light-cured composite resin (SDR®, Dentsply Sirona, New York, PA, USA). Mice were randomly assigned to the pulp exposure with saline application group (normal group; n=3), the pulp exposure with AITC application group (pulpitis group; n=3) and pulp inflammation with selective Nav 1.7 blocker, ProTxII (selective Nav1.7 blocker, 1 μM, Tocris Bioscience, Bristol, United Kingdom), treatment group (ProTxII group; n=3). All experiments were performed to minimize animal used.
Using sodium pentobarbital (75 mg/kg), mice were anesthetized and mounted on a stereotaxic apparatus (Narishige Scientific Instrument Laboratory, Tokyo, Japan) following three-day post-injection. A craniotomy was performed on the cortex and the dura was resected. Exposed brain was removed by suctioning with an aspirator until the trigeminal nerve is exposed. For staining, exposed TG were directly applied and stained for 1h with hydrophilic voltage-sensitive dye (VSD) di-2-ANEPEQ (50 mg/ml in saline, Molecular Probes, Eugene, OR, US). After staining, the TG was kept moist with saline. To verify the effects of Nav1.7 inhibition, ProTxII directly applied on TGs for 30 min following recorded the AITC-induced pulpitis model. In optical study, groups were divided such as normal (saline application group), pulpitis + before (AITC application group), and pulpitis + after (pulpitis and ProTxII treatment in TG group).
Optical signals were recorded from the TG, the origin of the mandibular branch (V3 ) nerve, activated orthodromically by stimulation of the mice whisker areas with stimulator electrode. Electrical stimulation (200 ms delay, 1 ms pulse width, 3 s interstimulus intervals, 0.1-1 mA intensity) was delivered via the needle stimulating electrode. Activated area and amplitude were analyzed upon electrical stimulation with 0.5 mA. The axis of the camera was positioned perpendicular to the surface of the TG and displayed the V3 in the image field. The change in fluorescence of VSD facilitated visualization of neuronal activity using a MiCAM02 system (BrainVision, Tokyo, Japan) consist with a high-resolution CCD camera (3.7 ms per frame maximum time). In vivo, optical imaging of eupnea typically required 10 acquisition sweeps and image acquisition was triggered by an electrocardiogram using stimulus / non-stimulus subtraction method. Ten consecutive images in response to the electrical stimulation of whisker areas were averaged to reduce background noise and artifacts. Image acquisition was triggered by an electrical stimulus and the magnification was provided by a 1 × objective and a 0.63 × projection lens (Leica Microsystems Ltd., Wetzlar, Germany), which yielded a detector array of 192 × 128 pixels. Activated area was analyzed for each color image and the converted area of each captured image was assessed as the percentage; activated area / whole captured area × 100. The optical intensity and activated area were analyzed using MetaMorph software (Universal Imaging Co., PA, USA).
After the experiments, the mice were perfused and the extracted ipsilateral TG specimens for identify the expression levels of Nav1.7 due to AITC-induced inflammation pain. Longitudinal sections were performed using a cryostat (Microm HM 525; Thermo Scientific, Walldorf, Germany) and collected tissues were blocked (5% normal goat serum containing 0.1% Triton X-100) for 1 h. The sections were incubated with primary antibody of rabbit anti-Nav1.7 anti-serum (1:1,000; Cell Signaling Technology, MA, United States) and anti-goat NeuN (1:1,000; NOVUS, CO, United States) for overnight at 4°C and then reacted with Alexa Fluor 488 and 647 Anti-rabbit / goat IgG secondary antibodies (1:1,000; Jackson ImmunoResearch, PA, United States). Counterstaining was used with DAPI (4’,6-diamidino-2-phenylin-dole). Immunofluorescence images were obtained using an LSM700 confocal microscope (Zeiss, Oberkochen, Germany).
Targeted brain stem (SpVc; spinal trigeminal nucleus) at each group was collected and immediately frozen in liquid nitrogen. These tissues were homogenized in lysis buffer (PRO-PREP; Intron Biotechnology, Pyeongtaek, Korea) containing phosphatase and protease inhibitors (PhosSTOP; Roche, Mannheim, Germany). The lysates were centrifuged at 13,000 rpm for 15 min at 4°C to separate the supernatants and the proteins were separated by SDS-PAGE. It was transferred to a PVDF membrane (Merck Millipore, Darmstadt, Germany). The membrane was blocked with 5% skim milk for 1h and incubated with anti-cFos antibody (1:1,000; Abcam, Cambridge, United Kingdom), anti-TRPA1 antibody (1:1,000; Abcam, Cambridge, United Kingdom), anti-ERK (1:1,000; Cell Signaling, MA, USA), anti-phospho-ERK (1:1,000; Cell Signaling, MA, USA), anti-CRMP (1:1,000; Cell Signaling, MA, USA), antiphospho-CRMP (1:1,000; Cell Signaling, MA, USA), and β-actin antibody (1:10,000; Cell Signaling, MA, USA) overnight at 4°C. The chemiluminescent substrate (GE Healthcare, Little Chalfont, United Kingdom) was used to detect the specific protein. The blots were analyzed using the LAS system (LAS 4000; GE Healthcare, Little Chalfont, United Kingdom).
All experiment values were presented as mean±SEM. In optical imaging <Fig. 1>, an analysis of variance with a student’s t-test for post-hoc analysis was used to compare differences between paired samples in pulpitis pain and Nav1.7 inhibitor, ProTxII, for the optical data. One-way ANOVA followed by Dunnett’s multiple comparisons was conducted to compare the differences groups in the western blot analysis <Fig. 3>. Statistical analyses were used with IBM SPSS program (ver. 23.0; IBM Corp., Armonk, NY, USA) and p<0.05 were considered statistically significant.
To observe the relieving pulpitis pain, we recorded the neuronal activity by membrane potential changes using VSD imaging. We investigated the spatial and temporal properties of in vivo TG neuronal activity following whisker electrical stimulation. The VSD images showed a representative electrical stimulus evoked response in the TG by 0.5 mA whisker stimulation <Fig. 1A>. In compare with normal and pulpitis groups, broader and higher patterns of activation area was observed in pulpitis pain. The temporal changes of color-changed pixels were counted and illustrated in <Fig. 1B>. In the amplitude, pulpitis + before group of TGs showed the increased neuronal activity compared to the normal group (pulpitis + before: 299.83±88.15, normal: 100±45.81). To verify the relieve effect of pulpitis pain, we observed the spatiotemporal dynamics of neuronal activity after 30 min Nav1.7 inhibitor, ProTxII, application. In pulpitis + after group, the neuronal activation pixels in vivo TGs were significantly reduced as compared with before and after the treatment of Nav1.7 channel inhibition by ProTxII (pulpitis + after: 147.92±96.70) <Fig. 1C, 1D> showed the higher activation pattern of strong response in the TGs were analyzed by pseudo red and green color pixel in our sturdy. The results showed statistically significant difference compared to the activation color red and green of the normal and pulpitis groups (pulpitis + before: 524.40±58.43, normal: 100±14.94). To determine whether ProTxII induced different color changed pixels, we observed the activation red and green color was significantly reduced as compared before and after the application of ProTxII (pulpitis + after:175.77±30.64). ProTxII inhibited the hyper-excitatory neuronal activity by less ten over 30-40% of optical signaling density as compared with pulpitis pain. These results indicated the increased hyper neuronal activity in pulpitis pain could be effectively reduced by Nav1.7 channel inhibition.
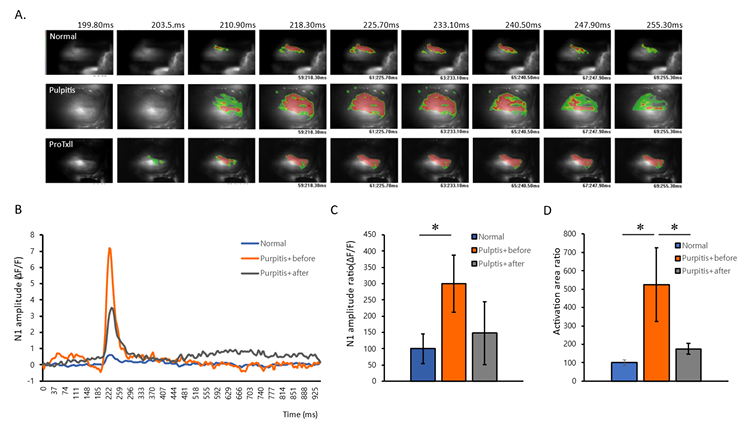
Fig. 1. Comparison of spatiotemporal neural activation in vivo optical recording. (A) Representative image of neural activation patterns in normal, pulpitis with before ProTxII treatment, and pulpitis after ProTxII treatment. (B) Comparison of time-dependent changes following stimulation in the electrical stimulation. (C) Comparison of neuronal amplitude. (D) Comparison of total neuronal activated areas. *p<0.05, n=3 per group. Data are presented as mean±SEM (Student t-tests; Normal vs Pulpitis+before, Pulpitis+before vs Pulpitis+after).
To investigate the cellular localization and the expression of Nav1.7, images from each group were randomly selected and subjected to immunohistochemistry. In the TG, large and small size of green-stained neurons are observed, with blue representing DAPI, which stains the nuclei of all cells. The double stained Nav1.7 positive neuronal cells exhibited with the bright green fluorescence. In pulpitis group, the highly activated Nav 1.7 channel was observed as compared with normal group. However, the treatment of ProTxII, Nav1.7 inhibitor, showed the inhibited the positive fluorescence neuronal cells. We found that the change in the protein expression of Nav1.7 in the trigeminal ganglia was consistent with that revealed by western blotting. Many trigeminal ganglion neurons were immune-positive for Nav1.7 in the acute pulpitis group and reduced the ProTxII treatment <Fig. 2>.
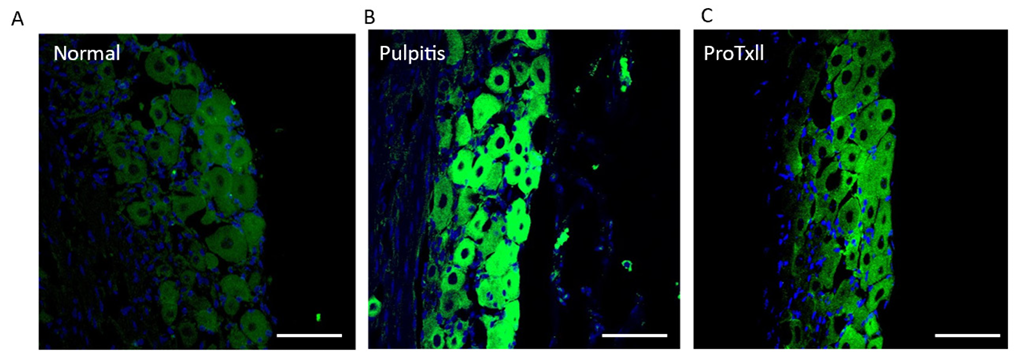
Fig. 2. Representative image of Nav1.7 expression in trigeminal ganglion. The Nav1.7-positive neurons were identified by immunohistochemistry. The expression of Nav1.7 was highly increased in the pulpitis group, but the expression of positive neurons was reduced in the ProTxII-treated groups. Scale bar: 50 μm.
To evaluate signaling pathway responses in alternation of Nav1.7 sodium channel activated by AITC-induced inflammation, we observed whether pulpitis induces the upregulation of pain signaling in the brain stem (SpVc) using western blots. We analyzed the expression levels of ERK, p-ERK, p-CRMP, CRMP, c-Fos and TRPA1 at the different groups <Fig. 3>. In brain stem, the expression level of p-ERK showed significantly increased in the pulpitis group (normal 1.04±0.04; pulpitis 2.00±0.20; ProTxII 1.60±0.15). And also, the c-Fos protein was tended to decreased after ProTxII exposure (normal 1.01±0.05; pulpitis 1.46±0.15; ProTxII 1.22±0.16). As the expression of TRPA1, the pulpitis group showed significantly upregulated in SpVc as compared to normal group, but downregulated the area which those given ProTxII in the expose (normal 1.27±0.31; pulpitis 1.63±0.05; ProTxII 1.03±0.20). The expression level of p-CRMP showed significantly increased in the pulpitis group (normal 0.94±0.06; pulpitis 3.08±0.87; ProTxII 2.34±1.14). In pulpitis group, the expression levels of p-ERK, p-CRMP, TRPA1, and c-Fos were significantly increased in the brain stem as compared with normal group. And also, the ProTxII prohibited inflammatory pulpitis pain showed tended to reduce the p-ERK and p-CRMP proteins as compared with pulpitis group. The results demonstrate the efficacy of ProTxII, a Nav1.7 inhibitor, in significantly reducing TRPA1 protein levels within the brain stem.
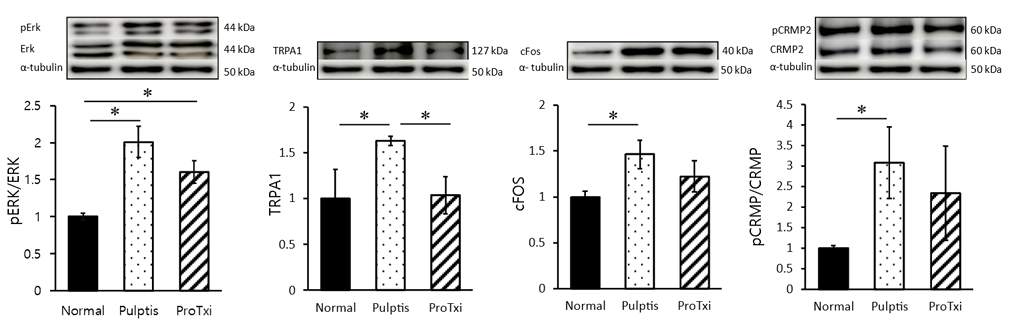
Fig. 3. Comparison of the changes in signaling molecules in each group. The signal changes of pERK/ERK increased significantly in pulpitis compared to normal, and this change was tended to reduce after ProTxII treatment. Between-group changes in TRPA1 and c-Fos. Increased TRPA1 and c-Fos changes were observed in pulpitis, and ProTxII decreased them in brain stem. The changes in pCRMP/CRMP were observed in pulpitis as induction of inflammation in SpVc area of brain stem. *p<0.05, n=3 per group. Data are presented as mean±SEM (one-way repeated measures ANOVA followed by Dunnett’ s multiple comparisons).
Pulpitis induced the activation of intracellular signaling pathways which lead to increase membrane excitability of voltage-gated sodium channels (VGSCs) [13]. In TG, an increased VGSC, involved in inflammatory pain, implicated the higher strength in synaptic lines and long-term potentiation with central sensitization [14,15]. In coincide with our previous in vitro study [11], we observed the neuronal hyper-excitability and the increased propagation of neuronal activity by using optical imaging of the in vivo TGs in a pulpitis model. In the context of pulpitis pain, the expression of Na channels, specifically Nav1.7, increased after pulp expose and associated with a Nav1.7 expression levels and degree of pulpitis pain. Also, the expression of Nav1.7 involved in the ectopic activity or abnormal firing of afferent nerves after inflammation [12]. Significantly, the Nav1.7 channel has been localized to sensory endings and act a major role in transmitting painful stimuli. After conditional knockout of Nav1.7 in the DRG, the pain hypersensitivity induced by nerve injury did not change [16]. Consist with the result of a previous study, the increase Nav1.7 was induced the lowering the threshold for spike initiation and producing hyper-excitability characterized by high frequency discharges in nociceptive TG neurons [11]. The Nav1.7 channel is sensitive to block by a nanomolar concentration of TTX and the Nav1.7 αsubunit caused to activate and inactivate inward currents with fast kinetics which were rapidly block by TTX in whole cell patch clamp techniques [17]. ProTxII showed effectively reduced the inflammatory induced hyperalgesia and neuronal hyper-excitability [11,18]. Moreover, we reconfirmed by in vivo optical recording that the treatment of ProTxII, a selective Nav1.7 blocker, on to the TGs in pulpitis model effectively suppressed the hyperpolarized activity after electrical stimulation. Nav1.7 channels have been found to be deployed at nociceptor nerve terminal, generator potentials occur in response to stimulation of the sensory nerve ending [19]. The presence of Nav1.7 serves the purpose of amplifying generator potentials and acts as a threshold channel, setting the gain in nociceptor [20]. The neuronal excitability regulated by VGSCs, which is spatially and temporally regulated and they possess distinct electrophysiological properties, may contribute in the pathophysiology of inflammatory pulpitis pain.
In the western blot analysis, we observed the SpVc, an important relay station for trigeminal nociceptive inputs from inflammation and tissue injury [21]. And also, SpVc nociceptive neurons had a crucial role in the transmission of nociceptive signals from the peripheral nervous system to the central nervous system following inflammatory injury [22]. ERK phosphorylation occurs in the trigeminal nervous system of rats with inflammatory pulpitis pain [12]. Moreover, the level of Nav1.7 significantly reduced by the treatment of an ERK antagonist in pulpitis induced rats. ERK phosphorylation is correlated with the involvement of trigeminal ganglionic Nav1.7 in hyperalgesia of inflamed temporomandibular joint. The depolarizing activation shift and a rapid Nav1.7 inactivation dependent on acute p-ERK inhibitions. As agreements, we also showed the expression level of p-ERK was tended to reduce following ProTxII treatment in the SpVc area. The action potential of dorsal root ganglion neurons by modulating Nav1.7 can regulate through the MEK/ERK pathway, the neurotrophic factors and pro-inflammatory cytokines [23]. p-ERK1 and c-Fos are wellknown markers of neuronal sensitization and are correlated with the degree of pain development [24]. The expression of c-Fos can indicate the physiologic activity to identify specific neuronal pathways in the brain and can be upregulated rapidly and dramatically after neural activity associated with pain [25]. In particular, the experimental tooth movement has been shown to induce the expression of c-Fos and TRPA1 in the trigeminal sensory complex, including the SpVc area [26]. As agreement, in this study, we the highly expressed c-Fos and TRPA1 was detected in pulpitis pain model of SpVc area. Thus, our western blot analysis indicates that the reduction of Nav1.7 expression can completely block peripheral nociceptive signaling mediated by VGSCs, thereby preventing the generation of spontaneous pain induced by inflammatory pulpitis pain. Central sensitization in inflammatory conditions is mediated by ongoing activities of primary afferents exposed to inflammatory irritants. Consequently, if the disease process is adequately controlled by sodium channel blockers, pain hypersensitivity may return to normal levels once the initiating event has healed. Nevertheless, the sodium channel blocker as an analgesic candidate is currently a relatively limited analgesic candidate in clinical trials due to its non-selectivity, relatively narrow therapeutic windows, and adverse side effects. The limitations of this study include the necessity to confirm the changes in Nav 1.7 in the brain stem and the small number of changes in proteins involved in sodium channel regulation. Future studies should investigate other signaling pathway which related with Nav 1.7 channel in the brain stem.
This study has focused on clarifying the role of the Nav1.7 channel to control nociceptive signal processing in pulpitis pain which confirmed orofacial pain behaviors related to the inflammatory response at our previous research [11] by in vivo electrophysiological recording.
1. The results of our study showed the hyper-excitatory neuronal activity in the TGs following dental pulp inflammation.
2. ProTxII, a selective Nav1.7 inhibitor, suppressed the increased neuronal excitability by membrane potential changes.
3. And the level of pain related signals was significantly reduced following ProTxII treatment. These findings suggest that the Nav1.7 channel may modulate nociceptive signal processing in pulpitis pain.
In conclusion, the control of Nav1.7 sodium channel was efficacious in animal models of pulpitis pain. This suggests that reduced Nav1.7 activation could be developed as a novel analgesic to relieve pulpitis pain in the brain stem. It may be useful for the management of inflammatory pain and for some aspects of neuropathic pain.
The author fully participated in the work performed and documented truthfully.
The author declared no conflicts of interest.
This study was supported by Dongseo University, ‘Dongseo Frontier Project’ Research Fund of 2023.
These experiments were approved by the Institutional Animal Care and Use Committee (IACUC approval No. 2021-0173) in the Laboratory Animal Facility at Yonsei University (Seoul, Republic of Korea).
Data can be obtained from the corresponding author.
None.
1. Aminoshariae A, Kulild JC, Donaldson M, Hersh EV. Evidence-based recommendations for analgesic efficacy to treat pain of endodontic origin: a systematic review of randomized controlled trials. J Am Dent Assoc 2016;147(10):826-39. https://doi.org/10.1016/j.adaj.2016.05.010
[DOI][PubMed]
2. Hossain MZ, Bakri MM, Yahya F, Ando H, Unno S, Kitagawa J. The role of transient receptor potential (TRP) channels in the transduction of dental pain. Int J Mol Sci 2019;20(3):526. https://doi.org/10.3390/ijms20030526
[DOI][PubMed][PMC]
3. Byers M, Narhi M. Dental injury models: experimental tools for understanding neuroinflammatory interactions and polymodal nociceptor functions. Crit Rev Oral Bio Med 1999;10(1):4-39. https://doi.org/10.1177/10454411990100010101
[DOI][PubMed]
4. Sharif-Naeini R. Role of mechanosensitive ion channels in the sensation of pain. J Neural Transm 2020;127(4):407-14. https://doi.org/10.1007/s00702-020-02182-2
[DOI][PubMed]
5. Tsumura M, Sobhan U, Muramatsu T, Sato M, Ichikawa H, Sahara Y, et al. TRPV1-mediated calcium signal couples with cannabinoid receptors and sodium–calcium exchangers in rat odontoblasts. Cell Calcium 2012;52(2):124-36. https://doi.org/10.1016/j.ceca.2012.05.002
[DOI][PubMed]
6. Lee KH, Lee BM, Park CK, Kim YH, Chung G. Ion channels involved in tooth pain. Int J Mol Sci 2019;20(9):2266. https://doi.org/10.3390/ijms20092266
[DOI][PubMed][PMC]
7. Cha M, Sallem I, Jang HW, Jung IY. Role of transient receptor potential vanilloid type 1 in the trigeminal ganglion and brain stem following dental pulp inflammation. Int Endod J 2020;53(1):62-71. https://doi.org/10.1111/iej.13204
[DOI][PubMed]
8. Afroz S, Arakaki R, Iwasa T, Oshima M, Hosoki M, Inoue M, et al. CGRP induces differential regulation of cytokines from satellite glial cells in trigeminal ganglia and orofacial nociception. Int J Mol Sci 2019;20(3):711. https://doi.org/10.3390/ijms20030711
[DOI][PubMed][PMC]
9. Meents JE, Bressan E, Sontag S, Foerster A, Hautvast P, Rösseler C, et al. The role of Nav1.7 in human nociceptors: insights from human induced pluripotent stem cell–derived sensory neurons of erythromelalgia patients. Pain 2019;160(6):1327-41. https://doi.org/10.1097/j.pain.0000000000001511
[DOI][PubMed][PMC]
10. Alvarez P, Levine JD. Antihyperalgesic effect of tetrodotoxin in rat models of persistent muscle pain. Neurosci 2015;311:499-507. https://doi.org/10.1016/j.neuroscience.2015.10.059
[DOI][PubMed][PMC]
11. Kwon MJ, Jung IY, Cha MH, Lee BH. Inhibition of the Nav1.7 channel in the trigeminal ganglion relieves pulpitis inflammatory pain. Front Pharmacol 2021;12:759730. https://doi.org/10.3389/fphar.2021.759730
[DOI][PubMed][PMC]
12. Sun S, Sun J, Jiang W, Wang W, Ni L. Nav1.7 via promotion of ERK in the trigeminal ganglion plays an important role in the induction of pulpitis inflammatory pain. Biomed Res Int 2019;1:1-11. https://doi.org/10.1155/2019/6973932
[DOI][PubMed][PMC]
13. Hameed S. Nav1.7 and Nav1.8: role in the pathophysiology of pain. Mol Pain 2019;15. https://doi.org/10.1177/1744806919858801
[DOI][PubMed][PMC]
14. Siqueira SRDT, Alves B, Malpartida H, Teixeira MJ, Siqueira JTT. Abnormal expression of voltage-gated sodium channels Nav1.7, Nav1.3 and Nav1.8 in trigeminal neuralgia. Neurosci 2009;164(2):573-7. https://doi.org/10.1016/j.neuroscience.2009.08.037
[DOI][PubMed]
15. Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Review Neurosci 2010;33:325-47. https://doi.org/10.1146/annurev-neuro-060909-153234
[DOI][PubMed]
16. Nassar MA, Levato A, Stirling LC, Wood JN. Neuropathic pain develops normally in mice lacking both Nav1.7 and Nav1.8. Mol Pain 2005;22(1)24. https://doi.org/10.1186/1744-8069-1-24
[DOI][PubMed][PMC]
17. Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J 1995;14(6):1084-90. https://doi.org/10.1002/j.14602075.1995.tb07091.x
[DOI][PubMed][PMC]
18. Emery EC, Luiz AP, Wood JN. Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin Ther Targets 2016;20(8):975-83. https://doi.org/10.1517/14728222.2016.1162295
[DOI][PubMed][PMC]
19. Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, et al. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Pro Natl Acad Sci 1997;94(4):1527-32. https://doi.org/10.1073/pnas.94.4.1527
[DOI][PubMed][PMC]
20. Rush AM, Cummins TR, Waxman SG. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol 2007;579(1):1-14. https://doi.org/10.1113/jphysiol.2006.121483
[DOI][PubMed][PMC]
21. Takeda M, Takahashi M, Matsumoto S. Suppression of neurokinin‐1 receptor in trigeminal ganglia attenuates central sensitization following inflammation. J Peripher Nerv Syst 2012;17(2):169-81. https://doi.org/10.1111/j.1529-8027.2012.00404.x
[DOI][PubMed]
22. Iwata K, Takeda M, Oh SB, Shinoda M. Neurophysiology of orofacial pain. Contemporary Oral Medicine, Springer International Publishing 2017;1-23. https://doi.org/10.1007/978-3-319-28100-1_8-2
[DOI]
23. Liu M, He F, Shao M, Li T, Wang L, Wang Y, Xu W. PACAP inhibition alleviates neuropathic pain by modulating Nav1.7 through the MAPK/ERK signaling pathway in a rat model of chronic constriction injury. Neuropeptides 2023;99(1):102327. https://doi.org/10.1016/j.npep.2023.102327
[DOI][PubMed]
24. Ji RR, Nackley A, Huh Y, Terrando N, Maixner W. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology 2018;129(2):343-66. https://doi.org/10.1097/ALN.0000000000002130
[DOI][PubMed][PMC]
25. Kato J, Wakisaka S, Kurisu K. Immunohistochemical changes in the distribution of nerve fibers in the periodontal ligament during an experimental tooth movement of the rat molar. Acta Anat 1996;157(1):53-62. https://doi.org/10.1159/000147866
[DOI][PubMed]
26. Reis CLB, Pingueiro-Okada EM, Luiz KG, Pedroso GL, Matsumoto MAN, de Menezes LM, et al. Orthodontic pain: c-Fos expression in rat brain nuclei after rapid maxillary expansion. J World Fed Orthod 2023;12(1):3-8. https://doi.org/10.1016/j.ejwf.2022.09.003
[DOI][PubMed]